Doi.org/10.57594/1055
Reynaga Cristhian1*, Prakash Manyu2, Prudente Isidoro2
1GlaxoSmithKline, Mexico
2ViiV Healthcare, Brentford, United Kingdom
*Autor de correspondencia: Reynaga Ortega Cristhian Daniel cristhian.d.reynaga@gsk.com Teléfono 55 4186 3911
Abstract:
Antiretroviral therapy has been shown to effectively suppress viral replication, with a progressive improvement of the safety and tolerability profile, allowing people living with HIV (PLWH) to live longer and have an improved number of comorbidity free years. This improvement has not been even; with a subset of PLWH requiring additional focus, including those who have experienced a sequential change of treatments due to the failure of the antiretroviral regimen, and due to development of antiretroviral-resistance mutations, toxicity, or the appearance of comorbidities that require complex treatments, with polypharmacy and drug-drug interactions (DDI). There is a clearly defined definition of heavily-treatment experienced (HTE) people with HIV although current DHHS guidance states that there is a subset of PLWH with limited treatment options due to multidrug resistance, intolerance, or drug-drug interactions. Effective management in this setting requires new fully active options, which would include those with new mechanisms of action (MoA). Fostemsavir is a new agent that acts blocking HIV-1 in entry process, binding to gp 120, preventing attachment, so there is no entry of the virus into the cell and the infection is not established. Since fostemsavir has been approved by COFEPRIS in Mexico, it becomes an option for HTE individuals. Here we will review the novel mechanism of action, pharmacologic profile, and present an update on fostemsavir clinical development.
Key words: Fostemsavir, Temsavir, Heavily treatment-experienced HIV patients, Multi drug resistance, CD4, gp120
Introduction
Effective options during HIV treatment are critical to ensure success in managing viremia and minimizing the potential risk of onset of comorbidities. For those individuals with limited options, the development of new agents with a new mechanism of action may be lifesaving. Current guidelines in the US and the EU state that HTE individuals are a subset of PLWH who possess limited remaining ART options due to resistance, intolerance, and potential interactions with concomitant medications. (1) However, there is no standardized definition at present, although this population is highly heterogeneous and can come from very different backgrounds in terms of treatment pathways and history. Challenges exist in this population in regard to the effective control of viral replication, a poor improvement of immune function (so called immune nonresponse, INR), toxicities (ART related and non-ART related) and, associated drug-drug interactions. (2) ART options and classes are essential for individuals with HTE, which also can offer improved tolerability and a limited cross-resistance profile to current ART classes. Consequently, these individuals require a more tailored individualized approach to their ART to address their specific needs.
It is important to consider the HTE population, as a much broader population than a multi-drug resistant, one as it can additionally include ART toxicity, comorbidities, and polypharmacy. In the US, HTE population has been estimated to range up to 14% (3, 4), whereas in Europe it is estimated to be 9% (5) with clear differences across Europe. These comprise both acquired drug resistance and transmitted drug resistance, can be applied to those individuals who have long lived with HIV and those who were recently infected.
Although the number of studies in the HTE setting have progressively decreased since 2010 (6), baseline parameters for participants in these studies have increased in terms of age, median and mean viral load (VL), CD4 cell count, median duration of prior ART and median number of prior ARVs. However, there are a number of newer drugs recently approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the management of multidrug-resistant HIV-1, which are ibalizumab, a monoclonal antibody that binds to the CD4 receptor after glycoprotein 120 attachment, (7) fostemsavir, also approved by the Federal Commission for the Protection against Sanitary Risks (COFEPRIS) in November 2022 in Mexico, and most recently, lenacapavir, a capsid inhibitor with a new mechanism of action (8). The aim of this article is to review the mechanism of action, pharmacology, and clinical development of fostemsavir.
Mechanism of Action and Pharmacology of Fostemsavir
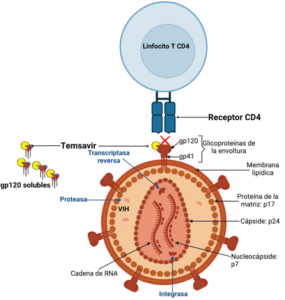
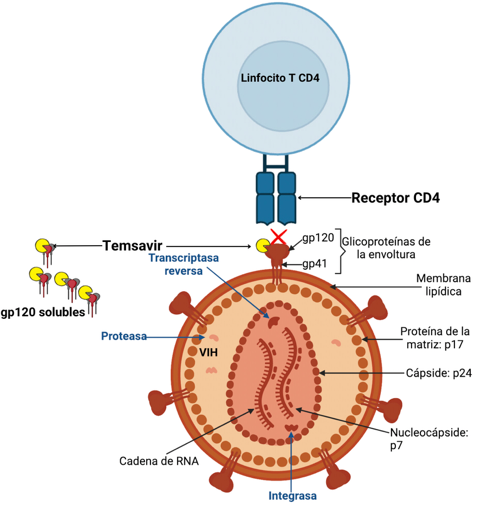
The entry of the HIV-1 virus into the host cell is a complex and sequential process comprising three stages: binding of glycoprotein 160 (gp160) to the CD4+ receptor, resulting in conformational changes in gp160 that allow binding to the co-receptor (CCR5 or CXCR4), which in turn induces larger conformational changes in gp160 that provoke fusion of viral and host cell membranes, which allows the HIV capsid structure to enter into the newly infected cell. (9). Gp160 is encoded by the env gene in the viral genome and after expression is subsequently cleaved by furin into glycoprotein 120 (gp120) and glycoprotein 41 (gp41) subunits. Three gp120 and three gp41 molecules combine into a heterodimer trimer to form the spike of the virus envelope. The structure of gp160 is non static, and the trimer oscillates between several states spontaneously: these are called closed, partially open and open states, with potentially intermediate states as well. Binding of the gp120 subunit to the CD4+ receptor only occurs in the open state. The virus entry process involves sequential structural rearrangements of both glycoproteins (10). In addition to facilitating this process, the interaction of gp120 with multiple host proteins can trigger cell signaling cascades that lead to cell toxicity and death, immune activation, inflammation, and organ and system dysfunction (11).
Temsavir (TMR) is the active metabolite of fostemsavir (FTR), the first-in-class inhibitor. It binds directly to gp120 near the CD4+ receptor binding site and locks gp120 in a closed state, preventing interaction of the glycoprotein with the CD4 receptor. In this way, fostemsavir prevents the first step of the virus entry process and the circulated unbound virus can subsequently be eliminated through host immune mechanisms (12).
Fostemsavir is administered orally every 12 hours in 600 mg extended-release tablets that must be swallowed whole without crushing, splitting, or chewing them. It is highly soluble in the gut and once it reaches the small intestine, it is converted to temsavir by alkaline phosphatase from epithelial cells on the luminal surface. Subsequently, temsavir is rapidly absorbed due to its high membrane permeability. The parent prodrug is not readily found in circulation (13).
TMR has a favorable pharmacological profile. It is primarily metabolized by the esterase-mediated hydrolysis pathway with contributions from the CYP450 3A4-mediated oxidative pathway. It is an inhibitor of the breast cancer resistance protein (BCRP) and organic anion transporter protein 1B1/3 (OATP1B1/3). It does not inhibit or induce the main CYP enzymes or uridine diphosphate glucuronosyltransferase. Consequently, interactions during co-administration of other available classes of antiretrovirals are not expected. Strong CYP3A inhibitors such as ritonavir or cobicistat increase TMR concentrations but no adjustment is required, as Cmax remains below levels associated with potential toxicity (QT prolongation). Moderate inducers of CYP3A, such as etravirine, decrease TMR concentrations, but dose modifications are also not required. Strong inducers of CYP3A, such as rifampicin, significantly reduce TMR concentrations, making co-administration contraindicated. Temsavir increases ethinyl estradiol concentrations, therefore a dose lower than 30 µg of ethinyl estradiol is recommended when administered concomitantly, while TMR has no impact on progestin. Since statins are substrates of OATP1B1/3 and BCRP, it is recommended to use statins at their lowest possible dose and monitor for adverse events. Elbasvir/grazoprevir is not recommended because inhibition of OATP1B1/3 increases concentrations and the risk of hepatotoxicity. Given that temsavir may cause QT prolongation at super-therapeutic concentrations, fostemsavir must be used with caution when co-administered with other drugs that may cause QT prolongation (14,15).
Clinical development of fostemsavir
A randomized, phase 2a study involving 50 HIV-1 subtype B-infected, naïve (70%) or experienced volunteers, with RNA HIV-1 levels >5000 copies/mL and CD4+ cells >200/mm3, evaluated the antiviral activity, pharmacokinetics, and safety of monotherapy with fostemsavir at different doses, from 600 mg to 1200 mg, once or twice daily with and without ritonavir in 5 treatment arms. After 8 days of treatment, the median maximum decrease in plasma HIV-1 RNA from baseline was between 1.21 and 1.73 log10 copies/mL. Short-term dosing of FTR ± RTV resulted in substantial decreases in plasma HIV-1 RNA levels in both naïve and experienced participants, all groups having a mean decrease in HIV-1 RNA of >1 log10. This drop in viral load is comparable to that seen with studies of monotherapy with other antiretrovirals. Fostemsavir was well tolerated. Although 78% of participants had >1 treatment-related adverse event, all were mild to moderate, with no grade 3 or 4 events reported, and there were no discontinuations. Co-administration of ritonavir produced only modest increases in drug exposure with no noticeable gains in viral load drop. These findings supported the subsequent studies with lower doses without boosters (16). Envelope genotypic analysis identified M426L as the primary substitution associated with a high baseline IC50 TMR and reduced antiviral response. However, M426L was also found in responders, therefore the role of this substitution in virologic response is unclear (17, 18).
AI438011 was a phase 2b, randomized, multicenter study that included 251 treatment-experienced patients with >1000 copies/mL of HIV-1 RNA and >50 cells/mm3 CD4+ T cells. Individuals were screened for susceptibility to TMR and only those with TMR IC50 <0.1µM (100nM) were enrolled. Five arms were examined, and participants received FTR at different doses (400 mg BID, 800 mg BID, 600 mg QD, and 1200 mg QD) and there was a control group with ritonavir-boosted atazanavir (ATV/r). To complete the regimen, all participants received standard doses of raltegravir (RAL) and tenofovir disoproxil fumarate (TDF) as a backbone. A monotherapy sub-study was also carried out in a subset of individuals randomized to FTR at different doses. In the monotherapy sub-study, FTR was active at all doses. Fostemsavir demonstrated mean plasma HIV-1 RNA decreases of 0.69 to 1.49 log10 copies/mL at day 7. At week 24, the 4 groups receiving fostemsavir showed similar efficacy compared to the group receiving ATV/r in the modified intention-to-treat analyses (69% to 80% vs. 75% respectively) and in the observed analysis (78% to 87% vs. 86% respectively). The median CD4+ T cell increase was 100 to 138 cells/mm3 in the FTR groups, while in the ATV/r group it was 103 cells/mm3. The incidence of virologic failure was similar between the FTR (16% to 26%) and ATV/r (18%) groups based on FDA snapshot algorithm. In the fostemsavir groups, 8 patients had a three-fold increase in FC IC50 from baseline, and four patients had emerging resistance to RAL. The inflection point for fostemsavir susceptibility was unclear and the clinical relevance of changes in susceptibility is not yet known. In the ATV/r group, there were no samples showing resistant mutations associated to ATV, TDF or RAL. Still, these data suggest that fostemsavir has a lower barrier to resistance than atazanavir. In general, fostemsavir was well tolerated in this study. Headache, often grade 1 and not dose related, was the most reported event (19). Recently at the 19th European AIDS Conference, the results related to inflammatory biomarkers were presented. Up to week 96, no consistent post-treatment differences were observed between the FTR and ATV/r groups in D-dimer, IP-10, IL-6, hs-CRP, or MCP-1 concentrations. Compared with ATV/r, FTR treatment resulted in a greater early and sustained decrease in concentrations of sCD14, a biomarker strongly associated with all-cause mortality in people living with HIV (20).
Although baseline substitutions at S375, M434, M426, and M475 positions were observed in viral samples obtained from 75/179 evaluable subjects in this study, no correlation was found between these substitutions and the number of subjects meeting the criteria for resistance testing. This suggests that, despite the variability of TMR susceptibility in this population (FC-IC50 range of 3.8-fold increase with S375S/N to >30,000-fold with M434I), virologic failure was not dependent on baseline FC-IC50 if subjects maintained some susceptibility to temsavir. In fact, 5 of 13 patients with a >3-fold increase in FC-IC50 and protocol derived viral failure from temsavir achieved viral suppression with the same regimen, including one participant with a 547-fold increase in FC-IC50. Thus, subjects may still respond to therapy despite baseline or emergent gp 120 substitutions. The impact of changes in temsavir’ s IC50 and emerging substitutions in HIV-1 gp120 will require further evaluations (21).
BRIGHTE study 5-year follow-up
BRIGHTE was a phase III, double-blind, placebo-controlled trial (with randomized and non-randomized cohorts) that included HTE patients with virologic failure defined by confirmed HIV-1 RNA >400 copies/mL. In the randomized cohort (RC), participants with one or two remaining classes of antiretrovirals with at least 1 fully active drug per class were allowed. In the non-randomized cohort (NRC), patients had no available classes of fully active drugs. Participants in the randomized cohort received monotherapy with fostemsavir 600 mg BID or placebo plus the current failed regimen for the first 8 days. Subsequently, all patients received fostemsavir 600 mg BID with an optimized background therapy (OBT) based on resistance tests and treatment history. In the non-randomized cohort, participants received fostemsavir with an OBT from day one (22).
A profile of the participants in the study revealed that more than 91% of people in both cohorts had previous exposure to NRTIs, NNRTIs, and PIs. In addition, approximately 75% of the randomized cohort and 95% of the non-randomized cohort had previously received INI therapy. In the non-randomized cohort, 100% of participants had exhausted NRTIs, PIs, and R5 antagonists, while 99% had exhausted all approved NNRTIs and INIs and 98% had exhausted fusion inhibitors. The most used components in the OBT were DTG, DRV, and TDF (23).
Of the 371 enrolled participants, 49% (133/272) of the randomized cohort and 23% (23/99) of the non-randomized cohort were still in the study at week 240 (June 2021). Eighty (22%) participants completed the study and switched to fostemsavir for commercial availability before week 240 (23).
Related to the primary outcome at day 8, the mean reduction in HIV-1 RNA was 0.79 log10 copies/mL in the fostemsavir group compared with 0.17 log10 copies/mL in the placebo group, with a statistically significant difference between treatments (22).
At week 96, the proportion of patients with <40 copies/mL of HIV-1 RNA in the RC in the intention-to-treat analysis was 60%, while at weeks 192 and 240 it was 53% and 45%, respectively. In the NC, the proportion of subjects with <40 copies/mL at week 96 was 37% in intention-to-treat analysis, while at weeks 192 and 240 it was 32% and 22%, respectively (Figure 1). After week 192, the results were altered by a lack of data related to the inability to attend study visits due to the COVID-19 pandemic. Virologic response rates were partially affected by a lack of data due to the COVID-19 pandemic and changes in OBT that were classified as treatment failures (24).
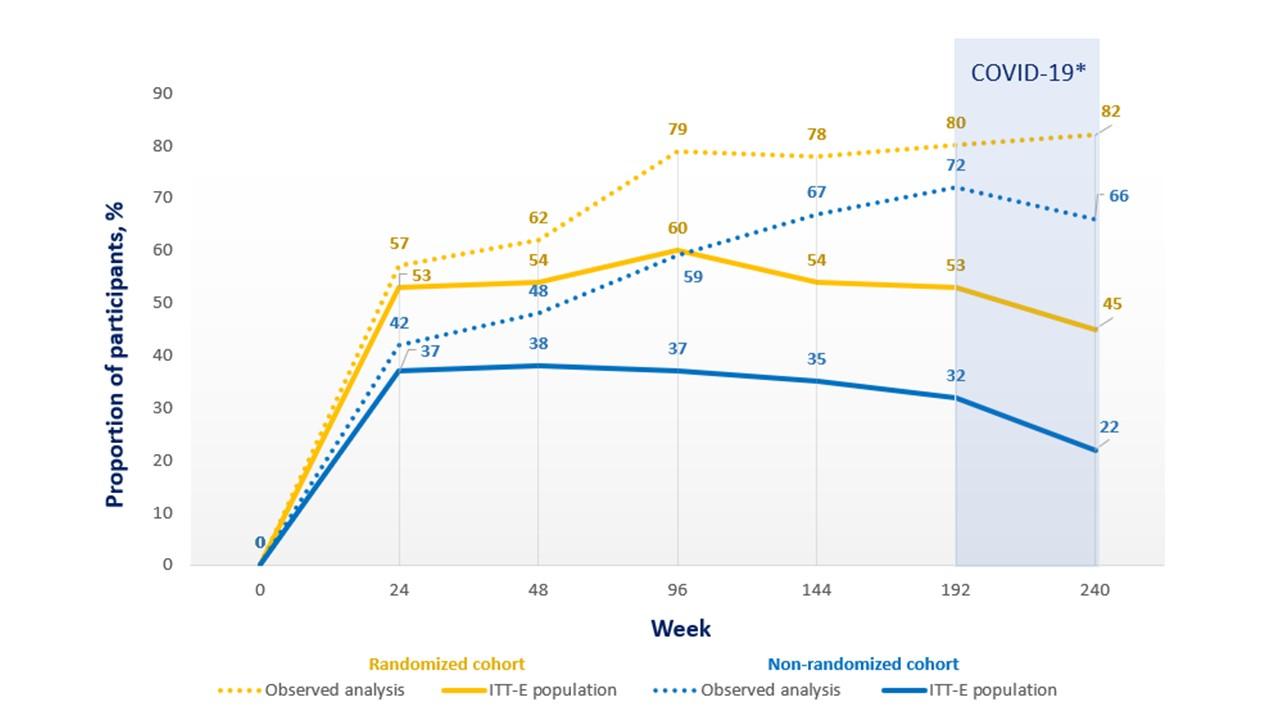
Figure 1. Virologic response through 240 weeks in the BRIGHTE study (adapted from: Aberg J, et al. IAS conference 2022. Poster EPB160)
In the RC, no difference in virologic response rates was observed among demographic subgroups (age, gender, race, and geographic region). The increase in CD4 count among demographic subgroups was also robust and continuous, with a mean change from baseline greater than 250 cells/mm3 at week 240. In those participants with baseline viral loads >100,000 copies/mL or CD4+ lymphocyte counts <20 cells/mm3, an increase in CD4 levels was also observed. 73/94 (78%) participants had a change in CD4+ T cell count from <200 to ≥200 cells/ mm3, and 22/33 (67%) had a change from <20 to ≥200 cells/mm3. Even patients who had not reached <40 copies/mL at week 240 had CD4+ cell increases (251 cells/mm3), reflecting immune improvement regardless of virologic response (25) (Figure 2) The mean CD4+/CD8+ ratio improved steadily at each time point evaluated, from 0.20 (SD, 0.24) at baseline to 0.60 (SD, 0.39) at week 240.
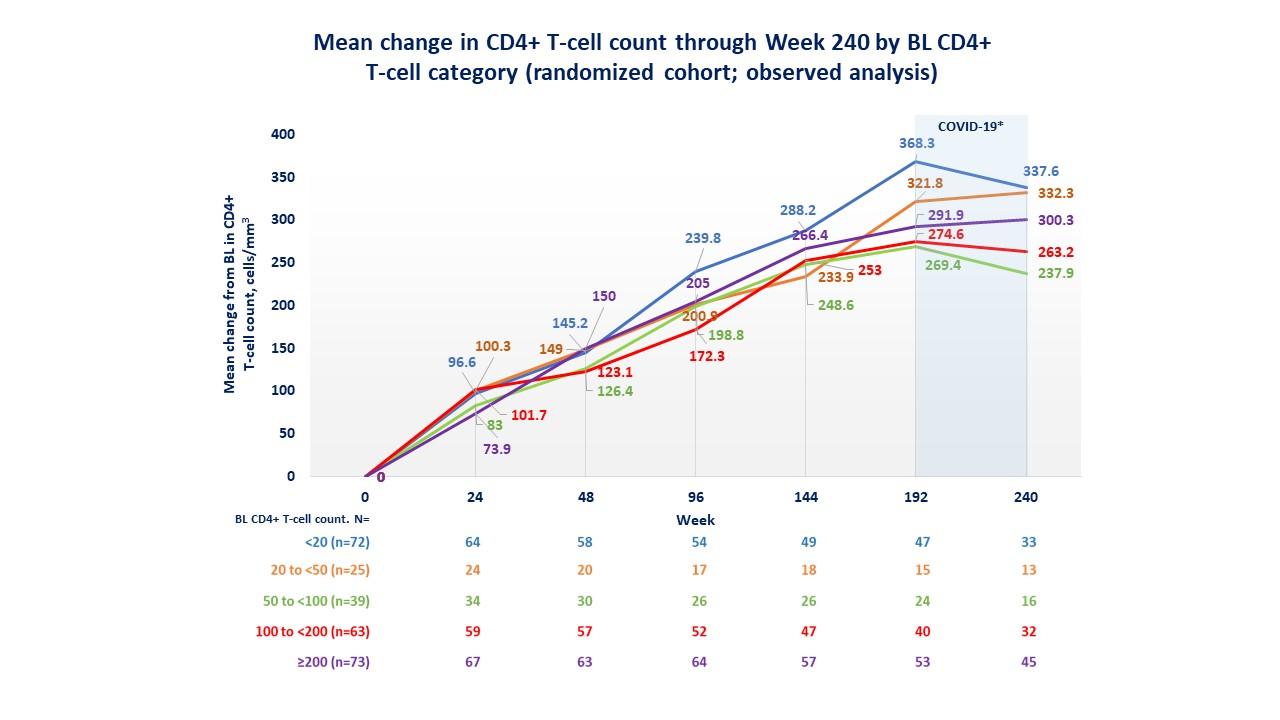
Figure 2. Sustained Immune Response Over 240 Weeks of the BRIGHTE Study (adapted from: Aberg J, et al. IAS conference 2022. Poster EPB160)
In the overall NRC, virologic response rates were similar across subgroups, although participants with poorer baseline disease status had lower response rates; however, interpretation was limited by the small number of participants. The median change in CD4+ T cell count from baseline was robust at week 240 for most subgroups, including among participants with baseline viral loads ≥100,000 copies/mL (176 [20, 383] cells/mm3) or baseline CD4+ T cell counts <20 cells/mm3 (216 [16, 1191] cells/mm3) (25).
In relation to coagulopathy, immune activation, and inflammation biomarkers there was a sustained improvement. The levels of D-dimer, sCD14, and sCD163 values decreased from baseline at week 240. These biomarkers have been linked to increased risk of mortality and non-AIDS-related comorbidities in people with HIV-1 (26).
During the 240-week follow-up, fostemsavir has shown a favorable safety and tolerability profile. Within both cohorts, 357/371 (96%) participants reported at least one AE, the most common being headache and nausea, and only 8% discontinued treatment mainly due to infections. Serious adverse events (SAEs) occurred more frequently in the non-randomized cohort. The majority of SAEs, AIDS-defining events and deaths were in participants with <200 CD4+ T cells/mm3. Deaths were primarily related to AIDS, acute infections, and non-AIDS malignancies. Eleven participants met the protocol-specified QTc-prolongation criteria and discontinued treatment. Immune reconstitution inflammatory syndrome (IRIS) was reported in 8 participants with one fatal event due to recurrent mycobacterial infection. None of them had cardiovascular symptoms and most of them (9/11) continued FTR treatment after the study discontinuation because they were enrolled in compassionate use program. Six participants became pregnant, all of them were from the randomized cohort and they were allowed to continue the FTR treatment. Of the 6 pregnancies, 3 resulted in normal births of healthy babies, 2 had complications (1 intrauterine growth retardation and 1 preterm birth) and 1 ended in elective abortion (27).
Up to week 240, the rate of protocol-defined virologic failure (PDVF) was 29% (80/272) in the randomized cohort and 54% (53/99) in the non-randomized cohort. In both cohorts, the majority of virologic failures occurred up to week 96, with an additional 17 and 4 participants in the randomized and non-randomized cohorts, respectively, with PDVF between week 96 and week 240. Among the evaluable participants, there was 30/80 in the randomized cohort and 33/53 with any emergent gp120 substitution of interest (changes at S375, M426, M434, M475). Since almost 50% of the PDVF participants in both cohorts had at least 1 polymorphism at these 4 sites at baseline and almost half of them had no substitutions of interest at week 240, the role of these mutations is not yet well defined (24).
Gartland et al (2020) described the results from genotypic and phenotypic analysis of baseline and on-treatment samples from participants meeting PDVF criteria trough week 96 with the aim of understanding the contribution to PDVF of polymorphism of interest and changes in viral susceptibility to temsavir. Gp120 emergent substitutions of interest that emerged during treatment were associated with increases in temsavir IC50 FC from baseline to PDVF, although there was a wide range of temsavir IC50 FCs observed in clinical samples with specific substitutions. Among randomized cohort participants with treatment-emergent gp120 substitutions at failure, median temsavir IC50 FC was 1,448, an increase of 511-fold above baseline IC50 FC, compared with a median IC50 FC of 3.1 and increase of 0.9-fold, respectively, for participants without treatment-emergent substitutions. Similar findings were observed in the non-randomized cohort. On the other hand, 52% of participants with PDVF in the RC and 25% in the NRC had no treatment-emergent gp120 substitutions of interest, and 55% and 29% in the RC and NRC, respectively, had a change in baseline temsavir IC50 FC within the variability assay (<3-fold). Therefore, phenotypic changes in temsavir susceptibility and treatment-emergent gp120 substitutions could not account for all cases of PDVF. Since there is no clear association between PDVF, reduced susceptibility to temsavir and treatment-emergent substitutions more data is needed to understand these substitutions effect (28).
Real-world evidence
Between July 2020 and September 2022, after fostemsavir was commercialized in the United States, 182 individuals initiated a fostemsavir based regimen in the OPERA cohort. 64% were viremic at baseline, of which 53% had a CD4+ cell count <350 cells/mm3. 36% were virologically suppressed and 44% of them had a CD4+ cell count <350 cells/mm3. Virologic control was maintained in most suppressed individuals, regardless of CD4 count. In contrast, 15 (52%) viremic patients with high CD4 and 10 (33%) viremic patients with low CD4 achieved virologic suppression. The greatest increases in CD4 were observed in suppressed individuals with low CD4 counts. Due to limitations of this cohort, more real-world data are required to understand why virologic response was low in viremic individuals. This real-world evidence supports the use of fostemsavir as a therapeutic option for HTE patients (29).
Conclusions
Fostemsavir is a new option for heavily treatment-experienced patients in Mexico, approved by COFEPRIS for people living with HIV-1 who have limited treatment options to build a fully active treatment regimen due to the development of resistance, toxicity, or intolerance, in combination with other medications. Fostemsavir offers a unique mechanism of action, with no cross-resistance to other entry inhibitors available in Mexico (maraviroc and enfuvirtide), as well as an acceptable pharmacologic profile, potency, barrier to resistance, and tolerability. The BRIGHTE study, with 5-year follow-up, supports the efficacy, safety, and durability of fostemsavir.
Abbreviations
PLWH, people living with HIV. HIV, human immunodeficiency virus. MoA, mechanism of action. gp, glycoprotein. RC, randomized cohort. NRC, non-randomized cohort. NRTI, nucleoside reverse transcriptase inhibitors. NNRTIs, non-nucleoside reverse transcriptase inhibitors. INI, integrase inhibitors. PI, protease inhibitors. CYP, cytochrome P450. QD, once a day. BID, twice a day. IC50, half maximum of the inhibitory concentration. IC50 FC, 50% inhibitory concentration fold change. Cmax, maximum concentration. hs-CRP, highly sensitive C-reactive protein. IP-10, inducible gamma interferon protein. MCP-1, monocyte chemotactic protein. TMR, temsavir. FTR, fostemsavir. DTG, dolutegravir. RAL, raltegravir. TDF, tenofovir disoproxil fumarate. ATV/r, atazanavir/ritonavir. AE, adverse event. SEA, serious adverse event. AIDS, acquired immunodeficiency syndrome. PDVF, protocol-defined virologic failure.
Bibliography
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv. Accessed January 2024.
- Hsu R, Fusco J, Henegar C, et al. Heavily treatment-experienced people living with HIV in the OPERA cohort: population characteristics and clinical outcomes. BMC Infectious Diseases (2023) 23:91
- Henegar C, Vannappagari V, Viswanathan S, DeKoven M, Clark A, Ackerman P, et al. Identifying heavily treatment-experienced patients in a large administrative claims database. In: International AIDS Society (IAS) conference on HIV science in Mexico City, Mexico, July 21–24, 2019. 2019
- Hsu R, Henegar C, Fusco J, Vannappagari V, Llamoso C, Lackey P, et al. Identifying heavily treatment-experienced patients in the OPERA Cohort [Poster THPEB044]. In: 22nd international AIDS conference, Amsterdam, Netherlands. 2018.
- Pelchen-Mattews A, Borges A, Reekie J, et al. Prevalence and outcomes for heavily treatment-experienced individuals living with human immunodeficiency virus in European cohort. J Acquir Immune Defic Syndr. 2021;87(2); 806-817.
- Aberg J, Mills A, Moreno S, et al. The evolution of clinical study design in heavily treatment-experienced persons with HIV: A critical review.
- Blair H. Ibalizumab: A review in multidrug-resistant HIV-1 infection. Drugs 2020 (80): 189-196
- US Food and Drug Administration. FDA approves new HIV drug for adults with limited treatment options. December 22, 2022. Available from: Accessed November https://www.fda.gov/news-events/press-announcements/fda-approves-new-hiv-drug-adults-limited-treatment-options. Accessed November 22, 2023
- Wilen C, Tilton J, Doms R. HIV: Cell binding and entry. Cold Spring Harb Perpect Med 2012; 2:a006866
- Chen B. Molecular Mechanism of HIV-1 entry. Trends in Microbiology 2019;27; 10:878–891.
- Yoon V, Fridkis-Hareli M, Munisamy S, et al. The gp120 molecule of HIV-1 and its interaction with T cells. Current Medicinal Chemistry, 2010;17:741–749
- Pancera M, Lai YT, Bylund T, et al. crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nature Chemical Biology 2017;13:1115–22
- Brown J, Chien C, Timmimns P, et al. Compartmental absortion modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J Pharm Sci 2013;102:1742-51
- Moore K, Post E, Mageau S, et al. Fostemsavir drug-drug interaction profile, an attachment inhibitor and oral prodrug of temsavir, for patients with multidrug-resistant- HIV-1 infection. International Workshop on clinical Pharmacology of HIV, Hepatitis, and other Antiviral Drugs Virtual 2020. Virtual; Oral Poster 14.
- Nwokolo N, Post E, Magee M, et al. Fostemsavir and ethinyl estradiol drug interaction: clinical application for co-administration. HIV Drug Therapy Glasgow 2020. Virtual; Poster P102
- Nettles R, Schurmann D, Zhu Li, et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis 2012;206:1002–11
- Zhou N, Nowicka-Sans B, McAuliffe B, et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother 2014;69:573–81
- Ray N, Hwang C, Healy M, et al. Prediction of virological response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J Acquir Immune Defic Syndr 2013;64:7–15
- Lalezari J, Brinson C, Echeverría J, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomized controlled trial. Lancet HIV 2015;2:e427–e437
- Wonderlich E, Prakash M, Clark A, et al. Early and durable reductions in soluble CD14 concentrations among treatment-experienced persons with HIV-1 through 96 weeks of fostemsavir treatment in a phase 2b clinical trial. 19th European AIDS conference 2023, Warsaw, Poland. Slides RA1.O4
- Lataillade M, Zhou N, Joshi S, et al. Viral drug resistance through 48 weeks in a pase 2b, randomized, controlled trial of HIV-1 attachment inhibitor prodrug, Fostemsavir. J Acquir Immune Defic Syndr 2018;77:299-307
- Kozal M, Aberg J, Pialoux G, et al. Fostemsavir in adults with multidrug-resistance HIV-1 infection. N Engl J Med 2020; 382:1232–43
- Lataillade M, Lalezari J, Kozal M, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced individuals: week 96 results of the phase 3 BRIGHTE study. Lancet HIV 2020;7: e740–51
- Aberg, J, Shepherd B, Wang M, et al. Efficacy and safety of fostemsavir plus optimized background therapy in heavily treatment-experienced adults with HIV-1: week 240 results of the phase 3 BRIGHTE study. 24th International AIDS Conference 2022 Virtual and Montreal, Canada. Poster EPB160
- Dyson A, Aberg J, Molina JM, et al. Durable efficacy and robust CD4+ T-cell count improvement observed across age, race, sex and geographic subgroups of heavily treatment-experienced people with multidrug-resistant HIV-1 after 240 weeks of fostemsavir treatment. IDWeek 2023; Boston, MA. Poster 365
- Castagna A, Mussini C, Llibre J, et al. Sustained improvements in biomarkers observed with fostemsavir in heavily treatment-experienced adults with multidrug-resistant HIV-1 from the phase 3 BRIGHTE study through week 240. 19th European AIDS Conference 2023, Warsaw, Poland. Poster eP.A.092
- Llibre J, Aberg J, Walmsley S, et al. Long-term safety and impact of immune recovery in heavily treatment-experienced adults receiving fostemsavir for up to 5 years in the BRIGHTE study. 19th European AIDS Conference 2023, Warsaw, Poland. Poster eP.A.093
- Gartland M, Cahn P, DeJesus E, et al. Week 96 genotypic and phenotypic results of the fostemsavir phase 3 BRIGHTE study in heavily treatment-experienced adults living with multidrug-resistant HIV-1. Antimicrobial Agents and Chemotherapy 2022. 1-15.
- Hsu R, Brunet L, Fusco J, et al. Fostemsavir use in the OPERA Cohort: Immunologic and virologic response. 19th European AIDS Conference 2023, Warsaw, Poland. Poster eP.A.068